HOT TOPICS IN PHARMACOGNOSY: Antibiotic Structures from the Past, and/or Novel Sources Now Being (Re)investigated

By David J. Newman, DPhil
INTRODUCTION (WITH SOME HISTORY)
The search for novel antibiotics by “interrogation” of the massive databases of compounds originally synthesized as part of the “conversion” of industrial chemical repositories to use compounds generated from combinatorial chemistry pipelines from the 1990s onwards has failed miserably (as documented by the GSK group in 20071 with further comments in 2015 by Blaskovich et al.2). Further documentation in publications from other groups is given in an excellent article by Scannell et al. in the December 2022 issue of Nature Reviews Drug Discovery.3 This article has an excellent “side bar box” showing how, from the title and contents, the 1930s “decision tool quality used by Domagk to discover and validate the sulfa drugs” beat the 1990 onwards decision tool quantity in antibacterial discovery, and no “synthetic chemistry substitute methodology” has risen since in industry.
Thus, what has become quite obvious (though whether the pharmaceutical industry has formally admitted it is an open question), is that the combinatorial chemistry techniques initially developed during those years have a well-defined place in the optimization of an active structure obtained from nature. An example of these techniques is shown in the later discussion on the oral antifungal agent ibrexafungerp.
LACK OF CONSISTENT ANTIBIOTIC DEVELOPMENT FUNDING BY INDUSTRY AND TO AN EXTENT BY GOVERNMENTS IN NORTH AMERICA AND THE EU?
The source(s) of funding for the initial discoveries, which today are almost without exception performed by academic groups either by themselves or as part of consortia (as in the EU), are from government grants. However, once potential agents are discovered and early promise is shown for a given structure, the current major problem is locating a viable and consistent funding source that is willing to cover the preclinical and clinical costs. These costs include the necessary infrastructure for production under GLP of adequate amounts to perform the necessary preclinical investigations, prior to consideration for clinical trials which might be stretched to Phase II using a GLP product, but GMP level is required for Phase III. Unfortunately, the resources for GLP production, let alone GMP, are not areas that are performed or funded for by academic labs. In addition, small biotech companies lack the infrastructure and funding that are necessary for further development.
One can argue, and I will, that this state of affairs has developed due to the perceived costs in “Pharmaceutical Industry C Suites” of maintaining industrial research groups investigating natural product sources in order to find a suitable agent that may lead to an anti-infective drug candidate. Unfortunately, the days when the leaders of major pharmaceutical houses were scientifically and/or medically trained are long gone.
However, there are some bright spots on the “source horizons,” particularly with academic consortia in the US / South America and the EU who have “attacked this problem from both directions”: the modification of what one might call “gray-haired active molecules” and the investigation of “unusual sources often insects and small mammals,” as sources of candidate molecules.
WHERE AND/OR WHY DO WE URGENTLY NEED NEW ANTIBIOTICS?
There are six bacteria identified by the WHO as the ESKAPE pathogens that currently do not have antibiotics consistently effective against them, due predominately from resistance profiles expanding as treatment continues. They are as follows: Enterococcus faecium, Staphylococcus aureus, Klebsiella pneumoniae, Acinetobacter baumannii, Pseudomonas aeruginosa and Enterobacter spp.
However, what is noteworthy in the list above, is the total absence of pathogenic fungi, even though infections by these agents, both old and new, are increasing as can be seen by the rise of Candida auris. This fungus is treatable by the echinocandins (which were first approved in the early 2000s), but strains resistant to these and the other two major classes of antifungal agents (polyenes and azoles) are now being recognized. Currently in the US and to a large extent in the EU, until very recently, only three NP-derived antifungal agents were approved in the last 20 years plus the odd synthetic azole. These were the three echinocandins: caspofungin in 2002, micafungin in 2005 and anidulafungin in 2006.
APPROVAL IN 2021 OF IBREXAFUNGERP
Then, very recently in the middle of 2021, ibrexafungerp [1-4]., a novel orally effective b-1,3-glucan synthesis inhibitor derived from the natural product enfumafungin, was approved by the FDA. The final route to the agent is shown in Figure 1. However, it took more years than the nominal 21 to achieve this agent, from the initial report by Schwartz et al.4 in 2000 to Apgar et al.5 published just before its approval by the FDA in the middle of 2021. The Apgar paper demonstrated the use of what can be considered combi-chem techniques to obtain subtly different structures at different stages of the optimization as shown in the referenced paper.
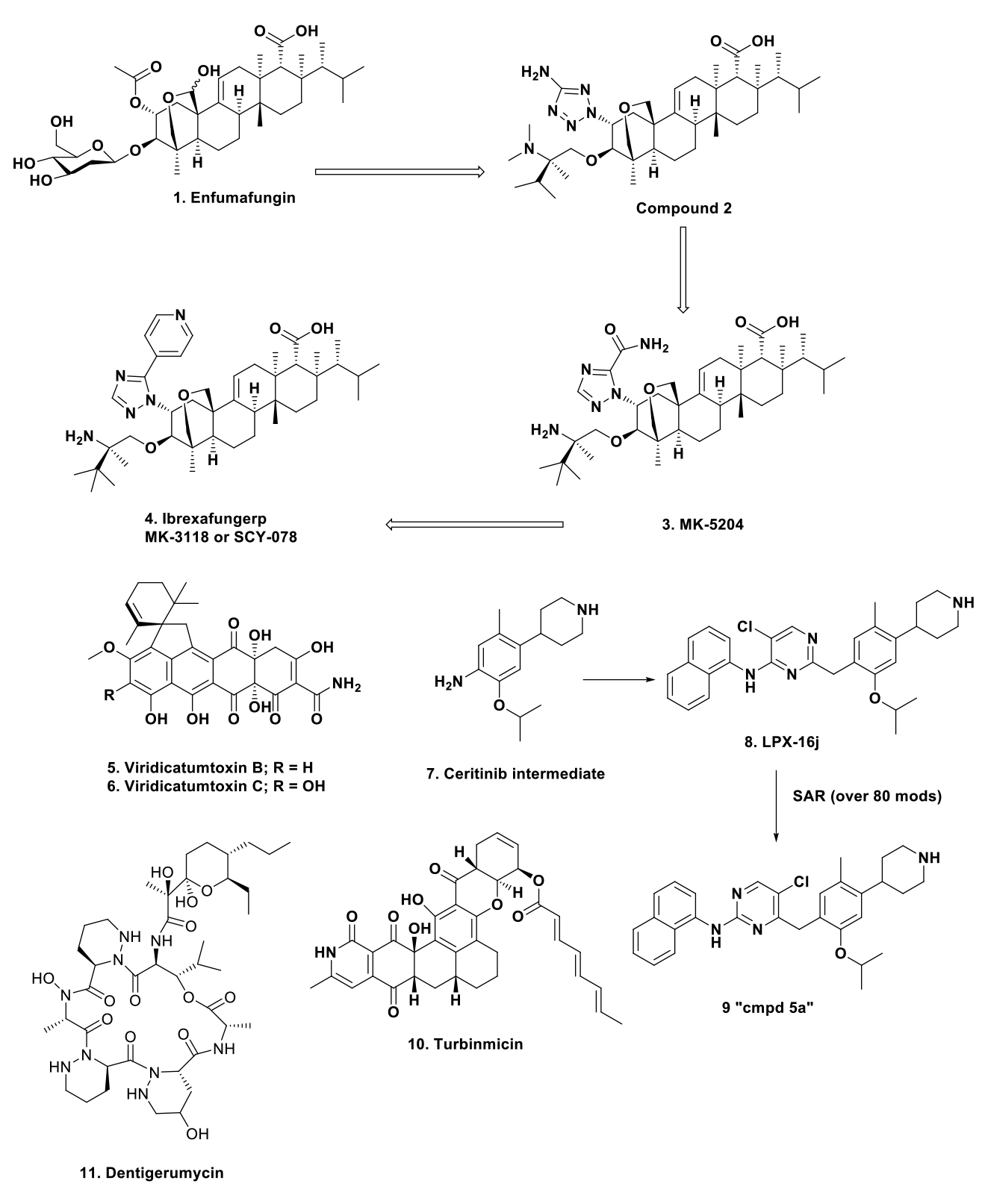
Figure 1. Structures (1 to 4 route to ibrexafungerp).
The reason why I used the term “nominal 21” above is that the first paper reporting the initial structure was by Pelaez et al.6 working in the then MSD laboratories in Madrid in 2000, where they isolated the compound from an endophytic Hormonema species isolated from leaves of Juniperus communis collected in Spain. The “sting is in the tail” of this paper is that only one Hormonema species produced this active agent, and by the time the article was published, the MSD scientists tested another 9000 fungal strains from their collection (from many different sources) plus another 8000 actinomycetes.
Unfortunately, today those biological resources are no longer available as the large, mainly US, pharmaceutical houses have “disposed” of their collections. They claim that they were not destroyed, but removal of these microbes and their associated high-cost support processes effectively means that they are no longer available.
CURRENT CHEMICAL MODIFICATION OF EXISTING NATURAL PRODUCT-DERIVED AGENTS
However, thanks to groups of talented chemists working with natural-product derived agents, there has been a significant amount of work reported as mentioned earlier, where combi-chem inspired processes have been applied to generate modifications of well-known antibiotic structures with the aim of overcoming microbial resistant processes. At the “risk” of pushing one of my own more recent editor-requested reviews,7 an open access article published in 2022 covered some of the potential molecules based on natural products that have used synthetic chemistry techniques (descendent of combi-chem perhaps?) to produce compounds with significant potential for future use.
BOGER’S VANCOMYCIN MODIFICATIONS
Of particular note, the vancomycin derivatives reported from the Boger laboratory in recent times have overcome the major vancomycin resistant microbes. In one of this group’s variations, a simple modification within the dipeptide buried in the overall structure (number 2 in reference 7 in the DJN review), yields a molecule that is active against previously resistant Gram-positive bacteria. The further extension of this excellent chemical foray led to the compound (number 3 or CBP-G3 in reference 7 in the DJN review). Further structural variations are mentioned in that review.
VIRIDICATUMTOXINS
These agents contain the basic tetracycline nucleus with “interesting” extensions between the A and B rings of the tetracycline. The original molecules viridicatumtoxin A and B were first reported in 1973 from Penicillium viridicatum, and the B variant was synthesized by Nicolaou in 20138 with a revision in 2014.9 A mixture of enantiomers was active against resistant strains of two of the ESKAPE pathogens (E. faecalis and MRSA) with activity also against E. faecium.
In contrast to the story on enfumafungin above, the biosynthetic gene cluster for these agents was found in a variety of fungi including A. nidulans, P. brasilianum and three other Aspergilli. The number of these toxins increased when it was realized from work by the Capon10 group in Queensland that a marine sourced endophytic Paecilomyces produced not only the A and B structures but also 4 more (C to F). Of the six compounds, B [5] and C [6] demonstrated a 15 to 40 fold difference in favor of antimicrobial activity versus cytotoxic activity in tested lines. The MoA of these compounds is “binding directly to the undecaprenyl pyrophosphate synthases (UPPS) of E. faecalis, S. aureus and E. coli with high affinity.” Since this target is an essential one for growth, it is a target worth considering, particularly since the BGC has been identified; targeted fermentation is a possibility to produce enough materials for larger-scale investigations. “Provided monies can be located!”
MODIFIED PYRIMIDINES AS POTENTIAL ANTI-TB CANDIDATES
A very recent paper by Li et al. in the Journal of Medicinal Chemistry11 demonstrated how, by utilizing intermediate [7] from the synthesis of ceritinib, the novel candidate LPX-16j [8] was synthesized. Ceritinib, though approved for use in anaplastic lymphoma via inhibition of the relevant kinase (ALK), was shown to have moderate inhibitory activity against the Mycobacterium tuberculosis strain H37Ra with an MIC of 9.0 µM. Using a suitable SAR regime, the preliminary lead compound LPX-16j [8] had an MIC of 5 µM against H37Ra but had a higher cytotoxicity against human cells with an MIC of 1-2 µM.
Using now standard combi-chemistry techniques, a long series of successive syntheses and biological testing, converted LPX-16j (basically a substituted pyrimidine) into compound 5a [9] which had a significantly lower toxicity, better oral activity and (MIC 0.5 µg/mL).
ACADEMIC GROUPS SOMETIMES WITH GOVERNMENT FUNDING (NIH, NSF, EU, ETC.) INVESTIGATING “UNUSUAL RESERVOIRS” OF NOVEL ANTIBIOTIC STRUCTURES
Though there are a number of groups that have pursued what could be considered in earlier days as “unusual sources,” three areas in particular have captured the “current scientific public eye.” These are not in chronological order but cover sea and land sources.
In the first to be discussed, a micromonospora species isolated from a sea squirt collected off the Florida Keys yielded the antifungal agent named as turbinmicin [10]. This agent turned out to be very active against Candida auris with an MIC of 0.25 0.5 µg/mL,12 and in a later paper Zhao et al.13 demonstrated the “killing mechanism” was inhibition of the biofilm vesicle production thus eliminating the matrix assembly.
The second source of novel antimicrobials against bacteria and/or fungi are the antibiotics expressed by microbes existing in the fungus gardens of fungus-growing ants. Granted the agents initially identified are protective agents against extraneous fungi that want to consume the ants’ food supplies, their “fungal gardens.” This work was pioneered by Clardy at Harvard Medical School in conjunction with Currie at the University of Wisconsin, with a publication in Nature Chemical Biology in 200914 identifying the novel antifungal agent dentigerumycin [11]. This work was part of a specific program funded by NIH in conjunction with other USG grant sources under a grant system that required participation with source country scientists, known by the acronym NCDDG. This system funded a very fruitful relationship with Monica Pupo and colleagues at the Pharmacy School at the University of Sao Paulo, Ribeirao Preto in Brazil, that permitted access to fauna in Brazil that has continued after the initial USG-sourced funding by utilizing a number of funding sources, including Brazilian. Two recent papers that aptly demonstrate the interplay between these researchers and their sources should be required reading in this field.15, 16
In addition to those mentioned above, another group, this time at the University of Oklahoma, have “pioneered” the study of what can be best described as the “microbiomes of roadkill.”17 Granted the Cichewicz group have been studying many other sources, but this one yielded some very interesting bacterial sourced agents. (In particular, a Pseudomonas and a Serratia species, both isolated from an opossum’s ear, inhibited drug-resistant C. albicans biofilm formation).
FINAL COMMENTS
Though some of the compounds shown above and those I have described in earlier columns may well have potential against microbial diseases that now are (or are becoming) major problems, the amount of funds available are nowhere near sufficient to develop a new antibiotic in spite of the requirement for such agents. As can be seen from comments above, significantly more than 20 years were necessary for the latest orally active antifungal agent to be approved. In earlier columns I have covered antibacterial agents uncovered by novel techniques by academic groups in the US, and a fair number have now had academic and/or small biotech companies synthesizing modified structures based on the originals, but to date, as far as I can tell, none have progressed to preclinical studies.

LITERATURE CITED
- Payne, D.J. et al. Drugs for bad bugs: confronting the challenges of antibacterial discovery. Nat. Rev. Drug Discov. 2007. 6: 29-40. doi.org/10.1038/nrd2201.
- Blaskovich, M.A.T. et al. Helping chemists discover new antibiotics. ACS Infect. Dis. 2015. 1: 285-287. doi.org/10.1021/acsinfecdis.5b00044.
- Scannell, J.W. et al. Predictive validity in drug discovery: what it is, why it matters and how to improve it. Nat. Rev. Drug. Discov. 2022. 21: 915-931. doi.org/10.1038/s41573-022-00552-x.
- Schwartz, R.E. et al. Isolation and structural determination of enfumafungin, a triterpene glycoside antifungal agent that is a specific inhibitor of glucan synthase. J. Amer. Chem. Soc. 2000. 122: 4882-4886. doi.org/10.1021/ja9944296.
- Apgar, J.M. et al. Ibrexafungerp. Bioorg. Med. Chem. Lett. 2021. 32: 127661. doi.org/10.1016/j.bmcl.2020.127661.
- Pelaez, F. et al. The discovery of enfumafungin, a novel antifungal compound produced by an endophytic Hormonema species biological activity and taxonomy of the producing organisms. System. Appl. Microbiol. 2000. 23: 333-343. doi.org/10.1016/S0723-2020(00)80062-4.
- Newman, D.J. Old and modern antibiotic structures with potential for today’s infections. ADMET & DMPK. 2022. 10: 131-146. doi.org/10.5599/admet.1272.
- Nicolaou, K.C. et al. Total synthesis and structural revision of viridicatumtoxin B. Angew. Chem. Int. Ed. 2013. 52: 8736-8741. doi.org/10.1002/anie.201304691.
- Nicolaou, K.C. et al. Total synthesis of viridicatumtoxin B and analogues thereof: strategy evolution, structural revision, and biological evaluation. J. Amer. Chem. Soc. 2014. 136: 12137-12160. doi.org/10.1021/ja506472u.
- Shang, A. et al. Fungal transformation of tetracycline antibiotics. J. Org. Chem. 2016. 81: 6186-6194. doi.org/10.1021/acs.joc.6b01272.
- Li, C. et al. Structure-activity relationship of novel pyrimidine derivatives with potent inhibitory activities against Mycobacterium tuberculosis. J. Med. Chem. 2003. doi.org/10.1021/acs.jmedchem.2c01647.
- Zhang, F. et al. A marine microbiome antifungal targets urgent-threat drug-resistant fungi. Science. 2020. 370: 974-978. doi.org/10.1126/science.abd6919.
- Zhao, M. et al. Turbinmicin inhibits Candida biofilm growth by disrupting fungal vesicle-mediated trafficking. J. Clin. Invest. 2021. 131: e145123. doi.org/10.1172/JCI145123.
- Oh, D.-C. et al. Dentigerumycin: a bacterial mediator of an ant-fungus symbiosis. Nature Chem. Biol. 2009. 5: 391-393. doi.org/10.1038/nchembio.159.
- Bae, M. et al. Chemical exchanges between multilateral symbionts. Org. Lett. 2021. 23: 1648-1652. doi.org/10.1021/acs.orglett.1c00068.
- Fukuda, T.T.H. et al. Specialized metabolites reveal evolutionary history and geographic dispersion of a multilateral symbiosis. ACS Cent. Sci. 2021. 7: 292-299. doi.org/10.1021/acscentsci.0c00978.
- Motley, J. L. et al. Opportunistic sampling of roadkill as an entry point to accessing natural products assembled by bacteria associated with non-anthropoidal mammalian microbiomes. J. Nat. Prod. 2017. 80: 598-608. doi.org/10.1021/acs.jnatprod.6b00772.
Vol XX Issue N
NEWSLETTER STAFF
Edward J. Kennelly, PhD
Editor In Chief
Patricia Carver, MA
Copyediting & Proofreading
Nancy Novick
Design & Production
Gordon Cragg, PhD
Mario Figueroa, PhD
Joshua Kellogg, PhD
Michael Mullowney, PhD
Guido Pauli, PhD
Patricia Van Skaik, MA, MLS
Jaclyn Winter, PhD
ASP Newsletter Committee
Contribution deadlines
Spring: Feb. 15; Summer: May 15 Fall: Aug. 15; Winter:Nov. 15
Please send information to
Edward J. Kennelly, PhD Editor In Chief,
ASP Newsletter
Department of Biological Sciences
Lehman College, CUNY
250 Bedford Park Blvd. West Bronx, NY 10468
718-960-1105
asp.newsletter@
lehman.cuny.edu
ISSN 2377-8520 (print) ISSN 2377-8547 (online)